0偐傜巒傔傞僄僋僜乕儉僨乕僞夝愅丂v1.0
栚師
仜偼偠傔偵
仜夝愅娐嫬傪惍偊傛偆
仜夝愅梡僣乕儖偺僀儞僗僩乕儖偲嶲徠僼傽僀儖偺擖庤
仜僄僋僜乕儉夝愅
仜僒儞僾儖僨乕僞傪梡偄偨夝愅
仜偍傢傝偵
壓偺恾偼丄DNA僔乕働儞僒乕1儔儞偁偨傝偱惗惉偝傟傞堚揱巕攝楍僨乕僞検偺擭師曄壔傪帵偟偰偄傑偡丅
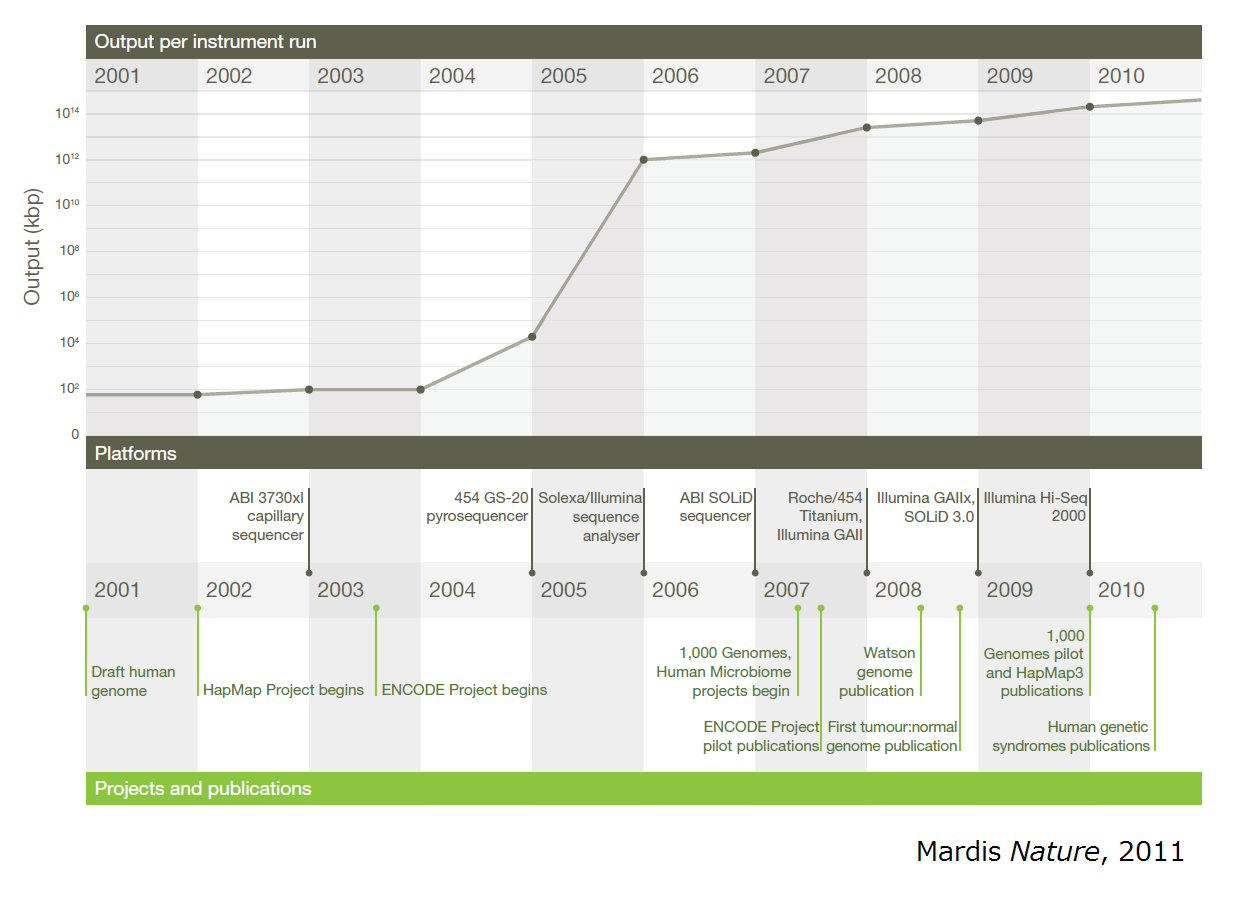
2005擭偐傜敋敪揑偵僨乕僞検偑憹偊偰偄傞偙偲偑傂偲栚偱暘偐傞偲巚偄傑偡偑丄偙傟偼丄師悽戙僔乕働儞僒乕媄弍偺奐敪偵傛傞傕偺偱偡丅
堦曽偱丄戝検偵惗惉偝傟偨僨乕僞傪夝愅偡傞偨傔偺媄弍偼丄愱栧惈偑崅傑偭偰偍傝丄昁梫側忣曬傪廤傔傛偆偲巚偭偰傕慜採抦幆傪梫偡傞傕偺偑懡偐偭偨傝丄擔杮岅偺忣曬偑彮側偐偭偨傝丄偲偄偆忬嫷偺偨傔丄傗傗晘嫃偑崅偔側偭偰偄傞姶偑偁傝傑偡丅
杮僐儞僥儞僣偱偼丄師悽戙僔乕働儞僒乕偺傾僾儕働乕僔儑儞偺堦偮偱偁傞乽僄僋僜乕儉夝愅乿偺曽朄偵偮偄偰丄巹帺恎丄僐儅儞僪儔僀儞傪巊偭偨夝愅偵偮偄偰偺抦幆偑杦偳側偄拞偐傜妛傫偩宱尡傪惗偐偟偰丄偱偒傞偩偗暘偐傝傗偡偔丄夝愢偟偨偄偲巚偄傑偡丅
師悽戙僔乕働儞僒乕偺棙梡曽朄偵偼條乆側傕偺偑偁傝傑偡偑丄偦偺墹摴偼丄傗偼傝僎僲儉偺攝楍夝愅偱偟傚偆丅偨偩丄慡僎僲儉乮僸僩偺応崌偍傛偦30壄墫婎懳乯傪夝愅偡傞偨傔偵偼丄傑偩傑偩帪娫傕偍嬥傕偐偐傞丄偲偄偆偺偑尰忬偱偡丅
偦偺拞偱丄旕忢偵岠棪偑偄偄曽朄偲偟偰棳峴偟偰偄傞偺偑丄乽僄僋僜乕儉夝愅乿偱偡丅偙偺曽朄偱偼丄愭偵Wet側幚尡偱僄僋僜儞乮Exon: 揮幨仺東栿傪宱偰丄嵟廔揑偵僞儞僷僋幙偺攝楍傪寛掕偡傞僎僲儉拞偺椞堟乯偺傒傪栐梾揑偵僉儍僾僠儍乕偟丄慖戰偝傟偰偒偨暘巕偺傒傪懳徾偲偟偰師悽戙僔乕働儞僒乕幚尡傪峴偄傑偡丅僄僋僜儞慡懱亖僄僋僜乕儉乮Exon+僊儕僔儍岅偺乽偡傋偰丒姰慡葌莻饒訓穫閻跀鰩珎蘯me偱Exome乯偺僒僀僘偼丄慡僎僲儉偺1-1.5%掱搙側偺偱丄偙傟偵傛偭偰丄僐僗僩偲帪娫偼戝暆偵嶍尭偝傟丄堦曽偱丄僞儞僷僋幙攝楍偵塭嬁傪媦傏偡廳梫側晹暘偺攝楍忣曬偼栐梾揑偵摼傞偙偲偑偱偒傑偡丅僄僋僜乕儉夝愅偺庡側栚揑偼丄曄堎偲幘姵側偳偺昞尰宆偺娭楢傪挷傋傞偙偲偱丄幚嵺偵丄偙偺曽朄偱丄婬側儊儞僨儖宆堚揱幘姵偺尨場堚揱巕傪摨掕偟偨丄偲偄偆榑暥偑師乆偲曬崘偝傟偰偄傑偡丅傑偨丄堚揱揑塭嬁傪庴偗傞傕偺偺丄偦偺宍幃偼傛傝暋嶨側幘姵乮惗妶廗姷昦丄帺屓柶塽幘姵丄惛恄恄宱幘姵側偳乯偺儕僗僋曄堎摨掕偵傕丄尰嵼恑峴宍偱妶梡偝傟偮偮偁傝傑偡丅
偝偰丄夝愅偵偼摉慠丄偦傟梡偺僜僼僩偑昁梫偵側偭偰偒傑偡偑丄WINDOWS OS偱偼忋庤偔摦偐側偄丄摦偐偦偆偲偡傞偲庤娫偑偐偐傞傕偺偑懡偄偺偱丄暿偺OS傪巊偆昁梫偑偁傝傑偡丅偙偙偱偼丄LINUX OS傪巊偆偙偲偵偟傑偡丅LINUX傪僀儞僗僩乕儖偡傞曽朄偲偟偰偼丄偡偭偐傝OS傪擖傟懼偊傞丄WINDOWS側偳懠偺OS偲椉曽摦偔傛偆偵偡傞丄側偳偺曽朄傕偁傝傑偡偑丄堦斣偍庤寉側偺偼丄壖憐PC傪摦偐偡偲偄偆曽朄偱偼側偄偐偲巚偄傑偡乮WINDOWS7偵偁傞Xp儌乕僪傕偙偺曽朄偱偡乯丅
壖憐PC傪摦偐偡僜僼僩僂僃傾傕偄傠偄傠偁傞偺偱偡偑丄偙偙偱偼丄傢傝偲恖婥丄柍彏丄64價僢僩偺壖憐儅僔儞偑摦偐偣傞乮婎杮揑偵丄師悽戙僔乕働儞僒乕夝愅偼64價僢僩儅僔儞偱側偄偲尩偟偄偱偡乯丄側偳偺棟桼偐傜丄VMware Player偲偄偆僜僼僩傪巊偭偰傒傑偡丅VMware Player偺僟僂儞儘乕僪偼偙偪傜乮http://www.vmware.com/jp/products/desktop_virtualization/player/overview乯偐傜丅搊榐偑昁梫偵側傝傑偡丅僀儞僗僩乕儔偺僟僂儞儘乕僪偑廔傢偭偨傜丄僂傿僓乕僪偵廬偭偰丄僀儞僗僩乕儖傪姰椆偝偣傑偟傚偆乮僨僼僅儖僩偺愝掕偱摿偵栤戣側偄偲巚偄傑偡乯丅
僀儞僗僩乕儖偑廔傢偭偨傜丄PC偺嵞婲摦偺屻丄VMware Player傪婲摦偟偰傒傑偡丅壓偺傛偆側僂傿儞僪僂偑弌偰偔傞偲巚偆偺偱丄乽怴婯壖憐儅僔儞偺嶌惉乿傪僋儕僢僋偟傑偡丅
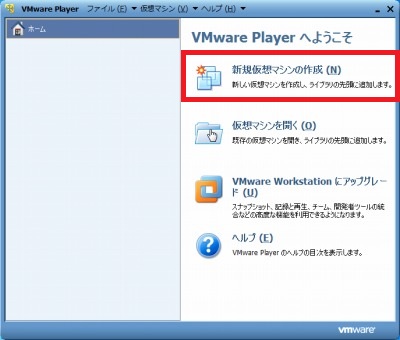
偡傞偲丄僎僗僩OS乮偙偙偱偼丄LINUX OS傪擖傟偨偄乯傪偳偺傛偆偵僀儞僗僩乕儖偡傞偐傪暦偐傟傞偺偱偡偑丄偙偺傑傑偱偼僀儞僗僩乕儖偱偒傑偣傫丅僀儞僗僩乕儔僨傿僗僋僀儊乕僕僼傽僀儖乮僀儞僗僩乕儖梡偺岝僨傿僗僋偺拞恎傪傑偲傔偨僼傽僀儖丄偲偄偆僀儊乕僕偱椙偄偐偲巚偄傑偡乯偲偄偆偺傪擖庤偡傞昁梫偑偁傝傑偡丅
傂偲偙偲偱LINUX OS偲偄偭偰傕丄偄傠偄傠側庬椶偑偁傞偺偱偡偑丄桳柤側偳偙傠偲偟偰偼丄Ubuntu丄Fedora偲偐偄偆柤慜偺傕偺偑偁傝傑偡丅偙偙偱偼丄Ubuntu儀乕僗偱丄僶僀僆僀儞僼僅儅僥傿僋僗夝愅梡偺僣乕儖偑嵟弶偐傜偁傞掱搙擖偭偰偄傞丄BioLinux偲偄偆僷僢働乕僕偺僀儊乕僕僼傽僀儖乮iso僼傽僀儖乯傪巊偭偰傒傑偡丅僼傽僀儖偺僟僂儞儘乕僪偼丄http://nebc.nerc.ac.uk/tools/bio-linux/bio-linux-6.0偐傜丄Download Now偵恑傫偱丄distro.ibiblio.org/bio-linux/iso/bio-linux-6-latest.iso.偲偄偆偲偙傠傪僋儕僢僋偡傟偽丄摿偵搊榐偟側偔偰傕擖庤偱偒傑偡丅
偝偰丄BioLinux偺iso僼傽僀儖偑庤偵擖偭偨傜丄VMware偺怴婯壖憐儅僔儞嶌惉夋柺偵栠偭偰丄僀儞僗僩乕儖僨傿僗僋僀儊乕僕僼傽僀儖偺偲偙傠偱丄愭傎偳擖庤偟偨僼傽僀儖傪慖戰偟偰丄師偵恑傒傑偡丅僎僗僩OS偼Linux丄僶乕僕儑儞偼Ubuntu64價僢僩傪慖戰偟丄懕偄偰奿擺応強側偳傪巜掕偟傑偡丅僨傿僗僋偺梕検偼丄彮偟梋桾傪帩偨偣偰偍偔偲偄偄偱偟傚偆丅儊儌儕側偳偼屻偐傜偱傕曄峏偱偒傑偡偑丄乽僴乕僪僂僃傾傪僇僗僞儅僀僘乿偺儃僞儞偐傜丄偙偺帪揰偱憹傗偟偰偍偔偙偲傕偱偒傑偡乮僗儉乕僘偵夝愅傪恑傔傞偵偼丄8G偖傜偄偼梸偟偄偲偙傠乯丅姰椆傪僋儕僢僋偡傞偲丄尵岅丄帪崗丄僉乕儃乕僪儗僀傾僂僩丄儐乕僓乕柤丄僷僗儚乕僪側偳傪慖戰丄擖椡偟偨屻丄帺摦偱僀儞僗僩乕儖偑恑傒傑偡丅
偙傟偱BioLinux偺僀儞僗僩乕儖偼姰椆偱偡偑丄偄傠偄傠偲VMware Player傪巊偄傗偡偔偡傞偨傔偵丄VMware Tools偲偄偆偺偑偁偭偨傎偆偑曋棙偱偡丅忋晹丄乽壖憐儅僔儞乿偺僞僽偵丄乽VMware-Tools偺僀儞僗僩乕儖乿偲偄偆崁栚偑偁傝丄偙偙傪墴偡偲丄僼僅儖僟偑奐偔偺偱丄傑偢偼拞偺僼傽僀儖傪僨僗僋僩僢僾偵堏摦偟偰傒傑偟傚偆丅
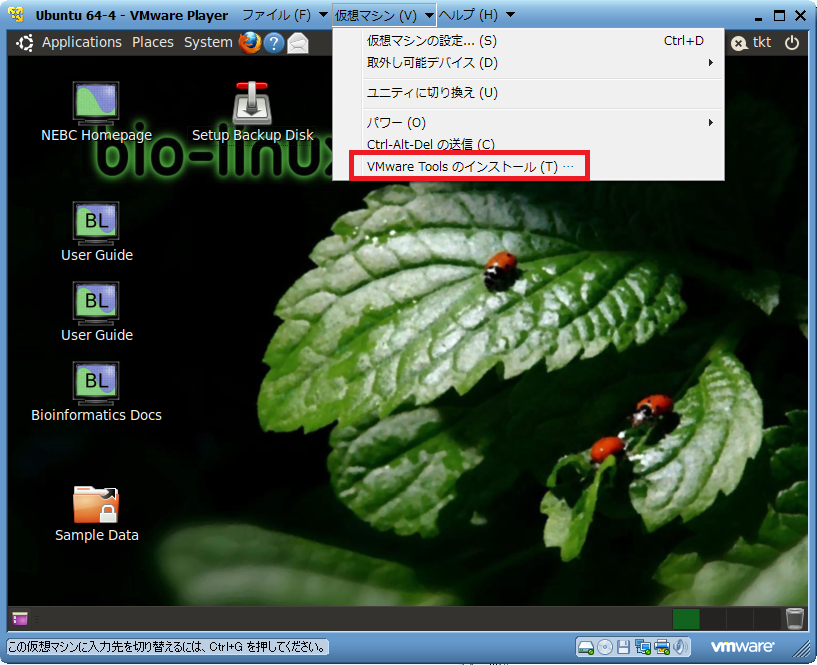
懕偄偰僀儞僗僩乕儖偵偲傝偐偐傞傢偗偱偡偑丄巆擮側偑傜丄WINDOWS偱傛偔傗傞傛偆偵丄僀儞僗僩乕儔傪僟僽儖僋儕僢僋偟丄僂傿僓乕僪偵廬偭偰乽師傊乿傪弴斣偵僋儕僢僋偡傟偽OK丄偲偄偆栿偵偼偄偒傑偣傫丅
僀儞僗僩乕儖偼丄僞乕儈僫儖乮WINDOWS偺僐儅儞僪僾儘儞僾僩偺傛偆側傕偺乯傪丄忋晹偺崟偄巐妏偺傾僀僐儞傪僋儕僢僋偟偰婲摦偟丄僐儅儞僪傪擖椡偟偰峴偆昁梫偑偁傝傑偡丅偙偺曈偑丄宱尡偑側偄恖偐傜偡傞偲晘嫃偑崅偄偺偱偡偑丄杦偳偺師悽戙僔乕働儞僒乕夝愅僣乕儖偼丄僞乕儈僫儖偐傜偺僐儅儞僪擖椡偱摦偐偡昁梫偑偁傞偺偱丄彮偟偢偮姷傟偰偄偒傑偟傚偆丅
僞乕儈僫儖傪婲摦偡傞偲丄壓偺傛偆側夋柺偑弌偰偒傑偡偑丄偙傟偼丄帺暘偑扤偱崱偳偙偺応強偵偄傞偐傪帵偟偰偄傑偡丅偙偺応強傪乽僨傿儗僋僩儕乿偲昞尰偟傑偡乮WINDOWS偺僼僅儖僟偺傛偆側傕偺偱偡乯偑丄僞乕儈僫儖傪巊偭偨僐儅儞僪擖椡偱偼丄壗傕巜掕偟側偄偲崱偄傞僨傿儗僋僩儕乮僇儗儞僩僨傿儗僋僩儕乯偵偁傞僼傽僀儖傪埖偆偙偲偵側傞偺偱丄昿斏偵僨傿儗僋僩儕傪堏摦偡傞昁梫偑偁傝傑偡丅偦偺偨傔偺僐儅儞僪偑cd乮Change Directory偺堄乯偱偡丅偨偩丄栚揑抧偵偨偳傝拝偔偨傔偵偼丄帺暘偑偳偙側傜峴偗傞偺偐偑暘偐傜側偄偲晄曋偱偡丅偦偙偱丄傑偢偼ls乮LiSt偺堄乯偲懪偭偰丄Enter傪墴偟偰傒偰偔偩偝偄丅僇儗儞僩僨傿儗僋僩儕偵偁傞僼傽僀儖偲僨傿儗僋僩儕偺儕僗僩偑弌偰偒偨偐偲巚偄傑偡丅惵偄暥帤偼僨傿儗僋僩儕傪堄枴偟偰偄傞偺偱丄偙傟傜偺応強側傜丄捈偵堏摦偡傞偙偲偑偱偒傑偡丅愭傎偳丄VMware-Tools偺僀儞僗僩乕儖梡僼傽僀儖偼僨僗僋僩僢僾偵抲偄偨偺偱丄
cd Desktop
偲擖椡偟偰丄僨僗僋僩僢僾偵堏摦偟傑偡丅
偙偙偱丄壓偺曽偵壗偐弌偰偄傞暥帤傪尒傞偲丄Tar傪巊梡偟偰僀儞僗僩乕儔傪揥奐偟傠丄偲彂偄偰偁傞偺偱丄偦傟偵廬偄傑偡丅
tar偲偄偆偺偼丄僐儅儞僪柤偱傕偁傝傑偡偑丄僼傽僀儖傪傂偲傑偲傔偵偟偨傕偺偺奼挘巕偱傕偁傝傑偡乮偲傝偁偊偢偼zip傒偨偄側傕偺偲峫偊偰偔偩偝偄乯丅
偄傠偄傠側奼挘巕偺僼傽僀儖傪揥奐乮夝搥乯偡傞偨傔偺僐儅儞僪偼丄偙偪傜乮http://uguisu.skr.jp/Windows/tar.html乯偵傛偔傑偲傑偭偰偄傑偡丅婰嵹偝傟偰偄傞TAR宍幃偺夝搥朄偵廬偭偰丄
tar xvf [VMware Tools偺僼傽僀儖柤.tar]
偲擖椡偟偰夝搥偟傑偡乮[]偺拞偵偼丄幚嵺偺僼傽僀儖柤傪擖傟偰偔偩偝偄乯丅tar偲xvf丄xvf偲僼傽僀儖柤偺娫偵偼丄敿妏僗儁乕僗傪嬻偗傞昁梫偑偁傝傑偡丅彫暥帤戝暥帤偑堘偭偰傕擣幆偝傟側偔側傝傑偡丅偪側傒偵丄xvf偲敿妏僗儁乕僗偺屻偱丄僞僽傪2夞墴偡偲丄僇儗儞僩僨傿儗僋僩儕偵偁傞僼傽僀儖堦棗偑昞帵偝傟丄偝傜偵僞僽傪墴偡偲丄弴斣偵僼傽僀儖偑慖戰偝傟傑偡丅傑偨丄xvf V偲懪偭偰僞僽傪墴偡偲丄V偐傜巒傑傞僼傽僀儖偺丄懕偒偺柤慜偑帺摦擖椡偝傟傞偺偱丄妝偡傞偙偲偑偱偒傑偡丅
偝偰丄僼傽僀儖偑夝搥偝傟偨偲偙傠偱壓偺曽偵弌偰偄傞暥帤傪嵞傃尒傞偲丄vmware-install.pl傪幚峴偟傠丄偲彂偄偰偁傝傑偡丅
偙偺僼傽僀儖偼丄僨僗僋僩僢僾偱偼側偔丄夝搥偟偨僼僅儖僟偺拞偵偁傞偺偱丄偦偪傜偵堏摦偟傑偡丅
cd vmware-tools-distrib
師偵vmware-install.pl偺幚峴偱偡偑丄偦偺傑傑vmware-install.pl偲懪偭偰傕忋庤偔偄偒傑偣傫丅慜偵壗傕彂偐偢偵暥帤楍傪擖椡偡傞偲丄僷僗偑捠偭偰偄傞乮http://www.obenri.com/_operation/path_through.html乯僨傿儗僋僩儕偵偁傞幚峴僼傽僀儖偼摦偐偡偙偲偑偱偒傑偡偑乮cd偲偐tar傪擖椡偟偨帪偑丄偙傟偵摉偰偼傑傝傑偡乯丄偦偆偱側偄僐儅儞僪傗僼傽僀儖側偳傪幚峴偡傞偙偲偼偱偒側偄偺偱偡丅
vmware-install.pl傪幚峴偡傞偵偼丄崱偙偙偵偁傞vmware-install.pl傪幚峴偟傠丄偲僞乕儈僫儖偵柦椷偡傞昁梫偑偁傝傑偡丅偦偺偨傔偵偼丄
./vmware-install.pl
偲擖椡偟傑偡丅"."偼僇儗儞僩僨傿儗僋僩儕傪帵偡婰崋偱丄僇儗儞僩僨傿儗僋僩儕偵偁傞vmware-install.pl偲偄偆僼傽僀儖傪幚峴偟傠丄偲偄偆堄枴偵側傝傑偡丅
偝偰丄忋婰偺傛偆偵擖椡偡傞偲乧忋庤偔偄偒傑偣傫偹丅
壗傗傜丄re-run this program as the super user偲昞帵偝傟偰偄傑偡丅偙傟偼丄偁側偨偵偼偙偺僼傽僀儖傪幚峴偟偰僀儞僗僩乕儖偡傞尃尷偑側偄偺偱丄僗乕僷乕儐乕僓乕偲偟偰幚峴偟側偝偄丄偲偄偆偙偲傪堄枴偟偰偄傑偡丅僗乕僷乕儐乕僓乕偲偄偆偺偼丄傑偀柤慜傪尒偨偩偗偱壗偐惁偦偆側偺偼暘偐傞偲巚偄傑偡偑丄傾僪儈僯僗僩儗乕僞乕尃尷偺傛偆側傕偺偱偡丅僗乕僷乕儐乕僓乕偲偟偰幚峴偡傞偨傔偵偼丄
sudo ./vmware-install.pl
偲擖椡偟偰壓偝偄丅偦偺屻丄僷僗儚乕僪傪梫媮偝傟傞偺偱丄傾僇僂儞僩僷僗儚乕僪傪擖椡偡傞偲丄柍帠幚峴偡傞偙偲偑偱偒傑偡丅
傑偨丄忢偵僗乕僷乕儐乕僓乕偲偟偰僐儅儞僪傪幚峴偡傞偵偼丄
sudo su
偲擖椡偟偰丄僷僗儚乕僪傪擖傟偰偔偩偝偄丅
儐乕僓乕柤傕曄峏偝傟偨偐偲巚偄傑偡丅
偝偰丄VMware-Tools偺僀儞僗僩乕儖偵栠傝傑偡偑丄僼傽僀儖傪幚峴屻丄彑庤偵恑傓偺偵廬偭偰Enter傪墴偟偰偄偗偽戝忎晇偱偡丅
偙傟偱丄僎僗僩OS偲儂僗僩OS乮尦偐傜擖偭偰偄傞OS乯偺峴偒棃偑僗儉乕僘偵側偭偨傝丄僐僺儁偑嫟捠偟偰偱偒偨傝偡傞傛偆偵側傝傑偡乮忋庤偔偄偐側偄偲偒偼丄僞乕儈僫儖偱vmware-user偲擖椡偟偨屻丄壗搙偐僎僗僩偲儂僗僩偺娫偱僐僺儁傪帋偟偰傒偰偔偩偝偄乯丅
傑偨丄廳梫側婡擻偲偟偰丄嫟桳僼僅儖僟傪愝掕偡傞偙偲偑偱偒傞傛偆偵側傝傑偡丅
偙傟偵傛偭偰丄僎僗僩丄儂僗僩偺椉曽偺OS偱丄僗儉乕僘偵夝愅偟偨傝丄僼傽僀儖傪堏摦偟偨傝偡傞偙偲偑壜擻偵側傝傑偡丅
偙偙偱偼丄昁梫嵟掅尷偺偙偲偟偐婰偟傑偣傫偱偟偨偑丄UNIX/LINUX偺婎杮揑側僐儅儞僪丄僨傿儗僋僩儕峔憿丄愨懳僷僗偲憡懳僷僗側偳偺梡岅偵偮偄偰丄偁傞掱搙棟夝偑偁偭偨曽偑丄屻乆偺夝愅偑僗儉乕僘偵側傞偲巚偄傑偡偺偱丄揔媂丄挷傋偰偄偨偩偗傑偡偲岾偄偱偡丅
傑偨丄偩偄偨偄偺僐儅儞僪偼丄
[僐儅儞僪柤] -h
偲偐丄
[僐儅儞僪柤] --help
偱丄僿儖僾傪昞帵偝偣傞偙偲偑偱偒傑偡丅
嫟桳僼僅儖僟偺愝掕偲丄壖憐儅僔儞偺愝掕偼丄偄偢傟傕乽壖憐儅僔儞乿僞僽偺乽壖憐儅僔儞偺愝掕乿偱壜擻偱偡丅孞傝曉偟偵側傝傑偡偑丄師悽戙僔乕働儞僒乕夝愅偵偼丄偐側傝偺儅僔儞惈擻偑昁梫偲側傝傑偡丅僴乕僪僂僃傾僞僽偺偲偙傠偱丄儊儌儕偲僾儘僙僢僒偺愝掕傪偟偰偍偒傑偟傚偆乮儊儌儕偺曽偼丄曄峏偡傞偵偼Player傪偄偭偨傫廔椆偡傞昁梫偑偁傝傑偡乯丅儊儌儕偼丄杮懱偺儊儌儕傛傝偪傚偭偲彮側傔偖傜偄偵愝掕偡傞偺偑偄偄偲巚偄傑偡偑丄8G偖傜偄偼梸偟偄偲偙傠偱偡丅僾儘僙僢僒偼丄4僗儗僢僪埲忋偁傞偺側傜丄嵟戝偺4偵偟偰偍偔偺偑椙偄偲巚偄傑偡丅嫟桳僼僅儖僟偼丄僆僾僔儑儞僞僽偺曽偱丄乽忢偵桳岠乿偵偟偰丄乽捛壛乿儃僞儞偐傜丄僂傿僓乕僪傪巊偭偰愝掕偟傑偡丅愝掕偑嵪傫偩嫟桳僼僅儖僟偼丄僎僗僩懁偱偼丄乭/mnt/hgfs/[巜掕偟偨嫟桳僼僅儖僟柤]乭偺僨傿儗僋僩儕偵懚嵼偟偰偄傑偡丅
偝偰丄LINUX OS偺僀儞僗僩乕儖偑嵪傫偩偲偙傠偱丄師偼夝愅僣乕儖偺僀儞僗僩乕儖偵側傝傑偡丅
偦偺慜偵丄僄僋僜乕儉夝愅偺婎杮揑側棳傟偼丄
嘆Wet側幚尡偲堦師攝楍夝愅偵傛傞丄僔儑乕僩儕乕僪偺攝楍偲奺墫婎偺僋僆儕僥傿傪帵偟偨僼傽僀儖乮嵟傕堦斒揑側宍幃偼Fastq僼傽僀儖乯偺惗惉
嘇僔儑乕僩儕乕僪偺嶲徠攝楍傊偺儅僢僺儞僌
嘊嶲徠攝楍偲偺斾妑偵傛傞曄堎乮堦墫婎曄堎=Single-Nucleotide Variant: SNV丄寚幐/憓擖曄堎=Insertion/Deletion: Indel乯偺僐乕儖
嘋曄堎偺傾僲僥乕僔儑儞乮娭楢忣曬偺晅壛乯
偲偄偆姶偠偵側傞偺偱偡偑丄偙偙偱偼丄嘇-嘋偵偮偄偰偲傝偁偘傑偡丅
崱夞僀儞僗僩乕儖偡傞僣乕儖偺儕僗僩偼丄
丒BWA
丒GATK
丒SAMTools
丒Picard
丒snpEff
偺5庬椶偱丄
奺僗僥僢僾偵偍偄偰丄
嘇仺BWA
嘊仺GATK
嘋仺GATK偲snpEff
傪婎杮揑偵梡偄傑偡丅
傑偨丄SAMTools偲GATK傪條乆側僼傽僀儖偺憖嶌偵丄Picard傪廳暋儕乕僪偺彍嫀梡偄傑偡丅
Burrows-Wheeler Aligner偺棯丅儅僢僺儞僌偺嵺僊儍僢僾傪嫋偡偺偱丄Indel偺摨掕偵岦偄偰偄傞傛偆偱偡丅夝愅帪娫偼傢傝偲偐偐傝傑偡丅懡暘尰帪揰偱偼嵟傕巊傢傟偰偄傞儅僢僺儞僌梡偺僣乕儖丅
僀儞僗僩乕儖曽朄丗
http://sourceforge.net/projects/bio-bwa/files/
偐傜丄嵟怴偺僼傽僀儖乮2011/10寧帪揰偱bwa-0.5.9.tar.bz2乯傪僟僂儞儘乕僪丅
僟僂儞儘乕僪偟偨僼傽僀儖偑偁傞僨傿儗僋僩儕偵堏摦丅
http://uguisu.skr.jp/Windows/tar.html傪嶲峫偵丄
tar -jxf bwa-0.5.9.tar.bz2
偱夝搥丅
cd bwa-0.5.9
偱堏摦丅
偙偙偱丄
make
偲擖椡偡傞偲僀儞僗僩乕儖偑彑庤偵巒傑傝傑偡丅
cp bwa /usr/bin/bwa
偱丄幚峴僼傽僀儖偱偁傞bwa偲偄偆僶僀僫儕僼傽僀儖傪丄僷僗偑捠偭偰偄傞僨傿儗僋僩儕偵僐僺乕
bwa
偲擖椡偟偰丄僶乕僕儑儞傗巊偄曽偑弌偰偒偨傜僀儞僗僩乕儖惉岟偱偡丅
The Genome Analysis Toolkit偺棯丅偄傠傫側僣乕儖偺廤崌懱偱丄堦岥偵偼愢柧偱偒側偄偺偱丄http://www.broadinstitute.org/gsa/wiki/index.php/The_Genome_Analysis_Toolkit傪偛嶲徠壓偝偄丅崱夞偼丄曄堎偺僐乕儖丄傾僲僥乕僔儑儞傗儅僢僺儞僌偺僉儍儕僽儗乕僔儑儞側偳偵巊偄傑偡丅
僀儞僗僩乕儖曽朄丗
Broad Institute偺拞偺恖偱側偗傟偽丄
ftp://ftp.broadinstitute.org/pub/gsa/GenomeAnalysisTK/GenomeAnalysisTK-latest.tar.bz2偐傜僟僂儞儘乕僪丅
僟僂儞儘乕僪偟偨僼傽僀儖偑偁傞僨傿儗僋僩儕偵堏摦丅
tar -jxf GenomeAnalysisTK-latest.tar.bz2
偱夝搥丅
埲忋偱僀儞僗僩乕儖偼姰椆丅
偙偺僣乕儖偼丄JAVA偲偄偆尵岅偱彂偐傟偰偄偰丄
枅夞
java -jar Path/To/GenomeAnalysisTK.jar
偱婲摦偡傞偙偲偵側傝傑偡丅乮Path/To偲偄偆偺偼丄GenomeAnalysisTK.jar偑偁傞僨傿儗僋僩儕傊偺愨懳僷僗傪帵偟偰偄傑偡丅嶲徠僐儅儞僪傪彂偄偰偁傞僒僀僩側偳偱偼丄偙偆偄偆昞婰朄偑偝傟偰偄傞偙偲偑懡偄偺偱偡偑丄偦偺傑傑Path/To偲擖椡偟偰傕摦偒傑偣傫丅乯
SAMTools偺SAM偼Sequence Alignment/Map偺堄枴丅SAM僼僅乕儅僢僩偼丄僔儑乕僩儕乕僪傪嶲徠攝楍偵儅僢僺儞僌偟偨寢壥偺僼傽僀儖宍幃偲偟偰丄嵟傕堦斒揑側傕偺偱偡丅SAM傪僨乕僞埑弅偺偨傔偵僶僀僫儕壔偟偨宍幃偑BAM僼僅乕儅僢僩偱丄SAM仺BAM偺曄姺丄BAM偺僜乕僩偵傛傞梕検偺嶍尭丄僜乕僩偟偨BAM偺僀儞僨僢僋僗嶌惉側偳傕SAMTools偱峴偄傑偡丅傑偨丄曄堎偺僐乕儖傕偱偒傑偡偑丄嵟嬤偼GATK偺Genotyper偱傗偭偨曽偑惛搙偑崅偄丄偲偺偙偲偱丄偦偪傜偱傗傞偙偲偑憹偊偰偄傑偡丅
SAMTools偼傕偲偐傜BioLinux偵偼擖偭偰偄傑偡偑丄偄偪偍偆嵟怴斉偵抲偒姺偊偰偍偒傑偟傚偆丅
http://sourceforge.net/projects/samtools/files/
偐傜丄嵟怴偺僼傽僀儖乮2011/10寧帪揰偱samtools-0.1.18.tar.bz2乯傪僟僂儞儘乕僪丅
僟僂儞儘乕僪偟偨僼傽僀儖偑偁傞僨傿儗僋僩儕偵堏摦丅
tar -jxf samtools-0.1.18.tar.bz2
偱夝搥丅
cd samtools-0.1.18
偱堏摦丅
make
偱僀儞僗僩乕儖丅
cp samtools /usr/bin/samtools
偱僷僗偑捠偭偰偄傞僨傿儗僋僩儕偵僐僺乕丅
samtools
偱丄僶乕僕儑儞偑0.1.18偵側偭偰偄傟偽OK偱偡丅
傄偐乕傞偲撉傒傑偡丅側偤Picard偐偼傛偔抦傝傑偣傫丅SAM丄BAM僼傽僀儖傪憖嶌偡傞偨傔偺JAVA尵岅偱彂偐傟偨僣乕儖孮偱丄偙偙偱偼廳暋儕乕僪乮Pair end偺椉儕乕僪偲傕攝楍偑姰慡偵堦抳偟偰偄傞傕偺丅摿掕偺PCR嶻暔偑曃偭偰憹暆偝傟傞偙偲側偳偵傛偭偰惗偠丄僇僶儗僢僕偺偽傜偮偒傗丄曄堎僐乕儖偺僄儔乕偵偮側偑傞丅乯傪彍嫀偡傞栚揑偱巊偄傑偡丅
http://sourceforge.net/projects/picard/files
偐傜丄嵟怴偺僼傽僀儖乮2011/10寧帪揰偱picard-tools-1.53.zip乯傪僟僂儞儘乕僪丅
僟僂儞儘乕僪偟偨僼傽僀儖偑偁傞僨傿儗僋僩儕偵堏摦丅
unzip picard-tools-1.53.zip
偱夝搥乮Lhaplus偲偐偱傕偄偄偑乯丅
偙傟傕丄
java -jar Path/To/XXX.jar
偱婲摦偡傞偺偱丄埲忋偱僀儞僗僩乕儖姰椆丅
SNP偺Effect傪傾僲僥乕僔儑儞偲偟偰偮偗傞偨傔偺僣乕儖丅僀儞僩儘儞偐僄僋僜儞偐丄傾儈僲巁抲姺傪婲偙偡偐婲偙偝側偄偐丄側偳偺忣曬偑晅壛偱偒傑偡丅
http://snpeff.sourceforge.net/download.html偐傜丄
僐傾僾儘僌儔儉乮http://sourceforge.net/projects/snpeff/files/snpEff_v2_0_3_core.zip/download乯傪僟僂儞儘乕僪偟偰夝搥丅
僸僩偺僨乕僞儀乕僗乮2011/10寧帪揰偱嵟怴偼GRCh37.64丂http://sourceforge.net/projects/snpeff/files/databases/v2_0_3/snpEff_v2_0_3_GRCh37.64.zip乯傪僟僂儞儘乕僪偟偰夝搥丅
data偲偄偆僨傿儗僋僩儕偑偱偒傞偺偱丄snpEff_v2_0_3僨傿儗僋僩儕偺拞偵奿擺偟傑偡丅
GATK側偳摨條丄
java -jar Path/To/snpEff.jar
偱摦偐偟傑偡丅
僣乕儖偑懙偭偨偲偙傠偱丄偝偀夝愅丄偲峴偐側偄偲偙傠偑傕偳偐偟偄偱偡偑丄偦偺慜偵丄條乆側嶲徠僼傽僀儖乮僸僩僎僲儉偺嶲徠攝楍丄曄堎偺儕僗僩側偳乯偑昁梫偵側偭偰偔傞偺偱丄偦傟傜傪傂偲捠傝擖庤偟傑偟傚偆丅
偙偺僼傽僀儖傪擖庤偱偒傞僒僀僩偼暋悢偁傝傑偡偑丄Broad Institute偺FTP僒僀僩傪椺偲偟偰偁偘偰偍偒傑偡丅
僷僗儚乕僪傪梫媮偝傟傑偡偑丄嬻棑偺傑傑偱OK偱偡丅
偙偪傜偐傜丄bundle丄1.2丄b37偲恑傫偩偲偙傠偵偁傞丄
human_g1k_v37.fasta.gz
偑1000僎僲儉僾儘僕僃僋僩偱巊傢傟偨僸僩偺嶲徠攝楍
human_g1k_v37.fasta.fai.gz
偑丄偦偺僀儞僨僢僋僗僼傽僀儖偱偡丅
fasta偲偄偆偺偑丄堦斒揑側僎僲儉嶲徠攝楍偺彂幃偵側偭偰偄傑偡丅
傑偨丄偙偺僼傽僀儖偱偼丄奺愼怓懱偺斣崋傪
1, 2, 3乧偲偄偆晽偵昞婰偟偰偄傑偡偑丄
chr1, chr2, chr3乧偲偄偆彂幃偵側偭偰偄傞嶲徠僼傽僀儖傕偁傝傑偡丅
偙偺昞婰朄丄愼怓懱偺弴斣偲傕偵懙偭偰偄側偄偲丄夝愅偑忋庤偔偄偐側偄偲偙傠偑偱偰偔傞偺偱丄拲堄偑昁梫偱偡丅
乮http://www.broadinstitute.org/gsa/wiki/index.php/Contig_ordering傪偛嶲峫壓偝偄乯
忋婰僒僀僩偵偼丄屆偄僶乕僕儑儞偺僸僩僎僲儉嶲徠攝楍側偳傕偁傝傑偡丅
曄堎偺儕僗僩傕偄傠偄傠側僶乕僕儑儞偑偁傝傑偡丅
嵟怴偺傕偺偼丄dbSNP乮http://www.ncbi.nlm.nih.gov/projects/SNP/乯偺FTP僒僀僩偐傜僟僂儞儘乕僪偱偒傑偡乮偙偙偺偲偙傠丄1000僎僲儉偺僨乕僞偑弌偰偒偨偙偲偱昿夞偵峏怴偝傟偰偄傑偡乯丅僼傽僀儖偺徻嵶偵偮偄偰偼丄奺僨傿儗僋僩儕偺Readme僼傽僀儖傪偛嶲徠壓偝偄丅
傑偨丄僼傽僀儖偺僼僅乕儅僢僩偲偟偰偼丄崱偼vcf宍幃乮E儊乕儖僋儔僀傾儞僩側偳偱巊傢傟偰偄傞揹巕柤巋偺宍幃偲摨偠柤慜偱偡偑丄暿偺傕偺偱偡乯偑堦斒揑偵側偭偰偒偰偄傑偡丅
彮偟屆偄傕偺偱椙偗傟偽丄忋弎偺Broad Institute偺FTP僒僀僩偵傕丄
dbsnp_132.b37.vcf.gz
dbsnp_132.b37.vcf.idx.gz
偲偄偆曄堎儕僗僩偺僼傽僀儖偲丄偦偺僀儞僨僢僋僗僼傽僀儖偑偁傝傑偡丅
崱夞偼偙偺僼傽僀儖傪巊偭偰傒傞偙偲偵偟傑偡丅
傑偨丄抁偄Indel偑偁傞晹暘偼丄僔儑乕僩儕乕僪傪忋庤偔儅僢僺儞僌偡傞偙偲偑擄偟偔側傝傗偡偄偺偱偡偑丄偦偺栤戣揰傪夝寛偡傞曽朄偲偟偰丄婛抦偺Indel偺廃曈偱丄嵞傾儔僀儞儊儞僩傪峴偆丄偲偄偆曽朄偑偁傝傑偡丅偦偺偨傔偵丄Indel偩偗偺儕僗僩偑昁梫偵側傝傑偡丅
偙傟傕丄Broad Institute偺FTP僒僀僩偐傜
1000G_biallelic.indels.b37.vcf.gz
1000G_biallelic.indels.b37.vcf.idx.gz
傪僟僂儞儘乕僪偡傟偽丄1000僎僲儉僾儘僕僃僋僩偱尒偮偐偭偨Indel偺儕僗僩偑擖庤偱偒傑偡丅
傑偨丄dbSNP傗1000僎僲儉偺vcf僼傽僀儖偵偼丄偨偄偰偄INFO偺僼傿乕儖僪偵VC (Variant classification) 偺崁栚偑偁傝丄偙偪傜偵SNP偐INDEL偐偑彂偐傟偰偄傑偡丅
grep偲偄偆UNIX僐儅儞僪乮摿掕偺暥帤楍偑偁傞峴丄側偄峴偩偗傪敳偒弌偡偙偲側偳偑偱偒傞乯傪巊偭偨壓偺傛偆側僐儅儞僪
zcat [vcf僼傽僀儖偺柤慜.vcf.gz] | grep -e ^# -e INDEL > [Indel偩偗偺僼傽僀儖偺柤慜.vcf]
偱丄僟僂儞儘乕僪偟偨gz僼傽僀儖偐傜丄INDEL偺偁傞峴偩偗傪拪弌偟偨僼傽僀儖傪嶌惉偡傞偙偲傕偱偒傑偡丅乮"|"偼僷僀僾丄">"偼弌椡偺儕僟僀儗僋僩偲屇偽傟傞傕偺偱丄擖弌椡偺庴偗搉偟偵曋棙側昞婰朄偱偡丅乯
僄僋僜乕儉夝愅偺応崌丄僞乕僎僢僩椞堟偑奺僉僢僩偛偲偵寛傑偭偰偄傑偡丅
僨乕僞偺僋僆儕僥傿僐儞僩儘乕儖乮暯嬒僇僶儗僢僕丄5X埲忋偱撉傔偰偄傞椞堟偺妱崌側偳乯偺偨傔偵丄偙偺僼傽僀儖傕擖庤偟偰偍偒傑偟傚偆丅
僼傽僀儖偼丄僄僋僜乕儉僉儍僾僠儍乕梡偺僉僢僩偺丄奺儊乕僇乕僒僀僩偐傜擖庤偱偒傞偲巚偄傑偡偑丄
Illumina TruSeq偼丂https://icom.illumina.com/download/summary/Ue6ccqdHiEmCuHC3cBLpxQ
Agilent SureSelect偼丂https://earray.chem.agilent.com/earray/丂偺eArray偺僒僀僩偐傜丄"exon"偺僉乕儚乕僪偱扵偟偰傕傜偊傟偽丄僟僂儞儘乕僪偱偒傑偡乮偄偢傟傕搊榐偑昁梫偱偡乯丅
eArray偺僒僀僩偐傜僟僂儞儘乕僪偱偒傞SureSelect偺僼傽僀儖偺応崌偼丄chr1, chr2, chr3乧偲偄偆彂幃偵側偭偰偄傞偺偱丄忋偱僟僂儞儘乕僪偟偨Fasta僼傽僀儖側偳偲崌傢偣傞昁梫偑偁傝傑偡丅
偙偙偱偼丄sed偲偄偆僐儅儞僪傪巊偭偰廋惓偟偰傒傑偡丅
sed s/chr// [僄僋僜乕儉僞乕僎僢僩椞堟偺僼傽僀儖.bed] > [廋惓屻偺僄僋僜乕儉僞乕僎僢僩椞堟偺僼傽僀儖.bed]
sed偼丄s偱抲姺婡擻傪巊偆偙偲傪巜掕偟丄"sed s/懳徾暥帤楍/抲姺暥帤楍/ 僼傽僀儖柤"偲偄偆宍偱擖椡偡傞偲丄奺峴偺嵟弶偵弌偰偔傞懳徾暥帤楍乮偙偙偱偼chr乯偑丄抲姺暥帤楍乮偙偙偱偼丄壗傕暥帤偑側偄乯偱抲偒姺偊傜傟傑偡丅
傑偨丄嵟弶偺僿僢僟峴傕嶍彍偟偰偍偔昁梫偑偁傝傑偡偑丄偙傟偼丄晛捠偵僥僉僗僩僄僨傿僞傪巊偭偰傕傜偊偽偄偄偲巚偄傑偡丅
偙偺崁偼丄http://www.vlsci.org.au/lscc/exome-pipeline偺夝愅僷僀僾儔僀儞偑傛偔傑偲傑偭偰偄偨偺偱丄偦偪傜傪嶲峫偵嶌惉偝偣偰偄偨偩偒傑偟偨丅
偄傛偄傛夝愅偵偲傝偐偐傝傑偡偑丄偦偺慜偵丄夝愅傪僗儉乕僘偵恑傔傞偨傔偺僨傿儗僋僩儕峔憿傪嶌惉偟丄傑偨丄壗搙傕巊偆僨傿儗僋僩儕丄僼傽僀儖偺応強傪帠慜偵巜掕偟偰偍偒傑偡丅
僨傿儗僋僩儕峔憿偼偁偔傑偱傕堦椺偱偡偑丄壓偺恾偺傛偆側宍偵偟偰偍偔偲丄憖嶌偑偟傗偡偄偐偲巚偄傑偡丅
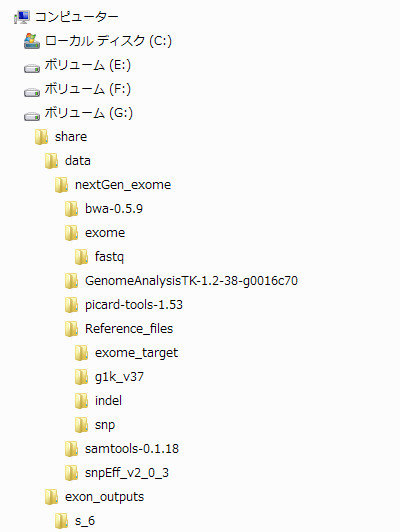
嫟桳僼僅儖僟壓偵data丄exon_output偺僨傿儗僋僩儕傪偮偔傝丄data偺拞偵nextGen_exome丄偦偺拞偵丄奺僣乕儖偺夝搥屻偺僨傿儗僋僩儕丄奺嶲徠僼傽僀儖偺僨傿儗僋僩儕偑擖偭偨Reference_files丄fastq僼傽僀儖偺僨傿儗僋僩儕偑擖偭偨exome偺僨傿儗僋僩儕傪丄偦傟偧傟堏摦丄嶌惉偟偰偄傑偡丅exon_output偺拞偵偼丄奺僾儘僕僃僋僩偺僨傿儗僋僩儕傪嶌惉偟偰偄傑偡丅
懕偄偰丄壗搙傕巊偆僨傿儗僋僩儕丄僼傽僀儖偺応強傪丄壓偺傛偆偵擖椡偟偰帠慜偵巜掕偟偰偍偒傑偡丅乮奺僣乕儖偺僶乕僕儑儞偑堘偆応崌偼丄偦傟偧傟廋惓偟偰偔偩偝偄丅乯
fastq_name=sequence.txt
i=6
#Fastq僼傽僀儖偺巜掕丅Illumina偺師悽戙僔乕働儞僒乕偺傾僂僩僾僢僩僼傽僀儖偼丄僨僼僅儖僩柤偑s_[儗乕儞柤]_[Pair end偺応崌丄"1"偐"2"]_sequence.txt偵側傝傑偡丅i=[儗乕儞柤]乮偙偙偱偼丄"6"偵偟偰偁傞乯偲巜掕偡傟偽丄僼傽僀儖柤傪曄峏偟側偔偰偄偄傛偆偵偟偰偄傑偡丅
fastq_dir=/mnt/hgfs/share/data/nextGen_exome/exome/fastq/
#Fastq僼傽僀儖偑偁傞僨傿儗僋僩儕傪巜掕丅
outdir=/mnt/hgfs/share/exon_outputs/s_6
#僼傽僀儖傪弌椡偡傞僨傿儗僋僩儕傪巜掕
reference=/mnt/hgfs/share/data/nextGen_exome/Reference_files/g1k_v37/human_g1k_v37.fasta
#嶲徠攝楍偺僼傽僀儖傪巜掕丅
dbSNP_file=/mnt/hgfs/share/data/nextGen_exome/Reference_files/snp/dbsnp_132.b37.vcf
#曄堎儕僗僩偺僼傽僀儖傪巜掕丅
indel_ref_file=/mnt/hgfs/share/data/nextGen_exome/Reference_files/indel/1000G_biallelic.indels.b37.vcf
#Indel偺僼傽僀儖傪巜掕丅
exon_capture_file=/mnt/hgfs/share/data/nextGen_exome/Reference_files/exome_target/SureSelect_All_Exon_G3362_with_annotation.hg19.bed
#僄僋僜乕儉僞乕僎僢僩偺僼傽僀儖傪巜掕丅
bwa_dir=/mnt/hgfs/share/data/nextGen_exome/bwa-0.5.9/
#BWA偺僶僀僫儕僼傽僀儖偑偁傞僨傿儗僋僩儕傪巜掕丅
samtools_dir=/mnt/hgfs/share/data/nextGen_exome/samtools-0.1.18
#samtools偺僶僀僫儕僼傽僀儖偑偁傞僨傿儗僋僩儕傪巜掕丅
gatk_dir=/mnt/hgfs/share/data/nextGen_exome/GenomeAnalysisTK-1.2-38-g0016c70/
#GATK偺僶僀僫儕僼傽僀儖偑偁傞僨傿儗僋僩儕傪巜掕丅
picard_dir=/mnt/hgfs/share/data/nextGen_exome/picard-tools-1.53/
#Picard偺僶僀僫儕僼傽僀儖偑偁傞僨傿儗僋僩儕傪巜掕丅
snpEff_dir=/mnt/hgfs/share/data/nextGen_exome/snpEff_v2_0_3/
#snpEff偺僶僀僫儕僼傽僀儖偑偁傞僨傿儗僋僩儕傪巜掕丅
#$偺屻偵丄愭傎偳擖椡偟偨嵍曈偺柤慜傪擖傟傞偲丄塃曈偺僨傿儗僋僩儕傪巜掕偡傞偙偲偑偱偒傑偡丅
#壓偺僐儅儞僪偱丄outdir偺壓偵丄搑拞偱弌椡偝傟傞僼傽僀儖傪奿擺偡傞偨傔偺僨傿儗僋僩儕傪嶌惉偟丄outdir/intermediate偵堏摦偟傑偡丅
mkdir $outdir/intermediate
mkdir $outdir/intermediate/tmp
mkdir $outdir/intermediate/coverage/
cd $outdir/intermediate
#BWA傪巊偭偰丄儅僢僺儞僌傪峴偄傑偡丅
#嶲徠Fasta僼傽僀儖偺Index傪丄index僐儅儞僪傪巊偭偰嶌惉偟傑偡丅
$bwa_dir/bwa index -p $reference -a bwtsw $reference
#嶲徠僼傽僀儖偛偲偵丄堦搙偩偗Index傪峴偆昁梫偑偁傝傑偡丅-p偱愙摢帿乮Prefix乯傪巜掕丅嶲徠Fasta僼傽僀儖偲摨偠偱OK丅-a偱傾儖僑儕僘儉傪巜掕丅傾儖僑儕僘儉偼bwtsw偲is偑偁傝傑偡偑丄僸僩慡僎僲儉偺傛偆側2GB埲忋偺僼傽僀儖偺応崌偼bwtsw丅僆僾僔儑儞偺屻偵丄敿妏僗儁乕僗傪嬻偗偰嶲徠Fasta僼傽僀儖傪巜掕丅
#偦傟偧傟偺Fastq僼傽僀儖偺僔儑乕僩儕乕僪傪丄aln僐儅儞僪偱嶲徠攝楍偵傾儔僀儞儊儞僩偟傑偡丅
$bwa_dir/bwa aln -t 4 $reference $fastq_dir"s_"$i"_1_"$fastq_name > "s_"$i"_1_.sai"
$bwa_dir/bwa aln -t 4 $reference $fastq_dir"s_"$i"_2_"$fastq_name > "s_"$i"_2_.sai"
#-t偼丄張棟偵巊偆僗儗僢僪悢丅僆僾僔儑儞偺屻丄Prexix偲Fastq僼傽僀儖傪巜掕丅Fastq僼傽僀儖傪弌椡偟偨Illumina僷僀僾儔僀儞僜僼僩僂僃傾偺僶乕僕儑儞偑1.3傛傝怴偟偄帪偼丄-I偺僆僾僔儑儞傪偮偗傑偡丅師偺僗僥僢僾偱擖椡偡傞僼傽僀儖偼奼挘巕偑.sai偱側偄偲捠傜側偄偺偱丄偦偺傛偆偵弌椡偟傑偡丅
#sampe偱丄Pair End偺儕乕僪偐傜SAM僼傽僀儖傪嶌惉偟傑偡乮Single end偺応崌偼samse乯丅
$bwa_dir/bwa sampe -P $reference "s_"$i"_1_.sai" "s_"$i"_2_.sai" $fastq_dir"s_"$i"_1_"$fastq_name $fastq_dir"s_"$i"_2_"$fastq_name -r "@RG\tID:01\tSM:s6\tPL:Illumina" > "s_"$i".sam"
#-P偱愭傎偳aln偟偨sai僼傽僀儖傪巜掕偟丄sai1 sai2 fastq1 fastq2偺弴斣偱暲傋傑偡丅-r偼丄Read group偺僿僢僟乕傪偮偗傞偨傔偺僆僾僔儑儞偱丄SM乮Sample乯偲PL乮Platform乯偑側偄偲GATK偑摦偒傑偣傫丅ID丄SM偼揔摉偱OK丅PL偼丄崱偺偲偙傠454, LS454, Illumina, Solid, ABI_Solid, CG偺偄偢傟偐偵偡傞昁梫偑偁傞傛偆偱偡丅弌椡偼.sam偱丅
#Samtools偺view僐儅儞僪偱SAM傪丄梕検偑彮側偄BAM偵曄姺丅
$samtools_dir/samtools view -bS "s_"$i".sam" > "s_"$i".bam"
#-b偱弌椡偑BAM偱偁傞偙偲丄-S偱擖椡偑SAM偱偁傞偙偲傪巜掕丅
rm "s_"$i".sam"
#偄傜側偔側偭偨SAM僼傽僀儖傪rm (remove) 偟傑偡丅
#Samtools偺sort僐儅儞僪傪巊偭偰丄BAM僼傽僀儖傪僜乕僩丅偙傟傪偡傞偲丄偝傜偵梕検偑尭傝傑偡丅
$samtools_dir/samtools sort "s_"$i".bam" "s_"$i"_sorted"
#僜乕僩偑廔傢偭偨傜丄BAM偵僀儞僨僢僋僗傪晅偗傞昁梫偑偁傝傑偡丅
$samtools_dir/samtools index "s_"$i"_sorted.bam"
#Picard偺MarkDuplicates偱丄廳暋儕乕僪傪彍嫀偟傑偡丅
java -Xmx2G -jar $picard_dir/MarkDuplicates.jar ASSUME_SORTED=true REMOVE_DUPLICATES=true INPUT="s_"$i"_sorted.bam" OUTPUT="s_"$i"_sorted_rem.bam" METRICS_FILE=duplicate VALIDATION_STRINGENCY=LENIENT
#廳暋儕乕僪彍嫀屻偺BAM偵僀儞僨僢僋僗嶌惉丅
$samtools_dir/samtools index "s_"$i"_sorted_rem.bam"
#Picard偺CreateSequenceDictionary偱丄Intarval僼傽僀儖偺嶌惉偵昁梫側丄嶲徠攝楍偺dict僼傽僀儖傪嶌惉丅
java -Xmx2G -jar $picard_dir/CreateSequenceDictionary.jar REFERENCE=$reference OUTPUT=/mnt/hgfs/share/data/nextGen_exome/Reference_files/g1k_v37/human_g1k_v37.dict
#GATK偺RealignerTargetCreator傪巊偭偰丄s_"$i"_sorted.bam偐傜嵞傾儔僀儞儊儞僩偟偨曽偑傛偝偦偆側Interval偺僼傽僀儖傪嶌惉丅
java -jar $gatk_dir/GenomeAnalysisTK.jar -T RealignerTargetCreator -R $reference -I "s_"$i"_sorted_rem.bam" --known $indel_ref_file -log intervals.log -L $exon_capture_file -o "s_"$i".intervals"
#GATK偺IndelRealigner偱丄愭傎偳挷傋偨Interval偺嵞傾儔僀儞儊儞僩傪巤峴丅
java -Djava.io.tmpdir=$outdir/intermediate/tmp -jar $gatk_dir/GenomeAnalysisTK.jar -T IndelRealigner -R $reference -I "s_"$i"_sorted_rem.bam" -log realigned.log -targetIntervals "s_"$i".intervals" -o "s_"$i"_realigned.bam"
#Picard偺FixMateInformation偱丄Pair end偺忣曬傪廋惓丅
java -Xmx2G -jar $picard_dir/FixMateInformation.jar INPUT="s_"$i"_realigned.bam" SO=coordinate VALIDATION_STRINGENCY=SILENT TMP_DIR=$outdir/intermediate/tmp
#嵞傾儔僀儞儊儞僩屻偺BAM僼傽僀儖偵僀儞僨僢僋僗嶌惉丅
$samtools_dir/samtools index "s_"$i"_realigned.bam"
#IndelRealigner偺帪偵偱偒偰偄偨僀儞僨僢僋僗僼傽僀儖傪嶍彍乮偙傟傪偟側偄偲丄師偺僗僥僢僾偑偆傑偔偄偐側偄乯丅
rm "s_"$i"_realigned.bai"
#gatk偺CountCovariates偲TableRecalibration傪巊偭偰丄嵞傾儔僀儞儊儞僩偟偨僼傽僀儖偺墫婎僋僆儕僥傿僗僐傾傪嵞寁嶼丅
java -jar $gatk_dir/GenomeAnalysisTK.jar -I "s_"$i"_realigned.bam" -R $reference --knownSites $dbSNP_file -nt 4 -T CountCovariates -l INFO -log recalc.log -recalFile recal_data.csv -cov ReadGroupCovariate -cov QualityScoreCovariate -cov CycleCovariate -cov DinucCovariate
java -jar $gatk_dir/GenomeAnalysisTK.jar -I "s_"$i"_realigned.bam" -R $reference -T TableRecalibration -l INFO -recalFile recal_data.csv -log new_qual.log --out "s_"$i"_realigned_alnRecal.bam"
#嵞傾儔僀儞儊儞僩丄嵞寁嶼屻偺BAM僼傽僀儖偵僀儞僨僢僋僗嶌惉丅
$samtools_dir/samtools index "s_"$i"_realigned_alnRecal.bam"
#GATK偺DepthOfCoverage偱僇僶儗僢僕偺寁嶼丅
java -jar $gatk_dir/GenomeAnalysisTK.jar -T DepthOfCoverage -R $reference -I "s_"$i"_realigned_alnRecal.bam" -o $outdir/intermediate/coverage/"s_"$i"_depth" -L $exon_capture_file -omitBaseOutput -ct 5 -ct 10 -ct 20 -ct 30
#-ct偱丄5X埲忋僇僶儗僢僕偺妱崌丄10X僇僶儗僢僕埲忋偺妱崌側偳傪昞帵偝偣傞偙偲偑偱偒傑偡丅-geneList偱丄僄僋僜儞偛偲丄堚揱巕偛偲側偳偺僇僶儗僢僕傪挷傋傞偙偲傕偱偒傑偡丅
#Bedtools偺coverageBed傪巊偆曽朄偼妱垽丅
#GATK偺UnifiedGenotyper傪巊偭偰丄SNV偺僐乕儖傪偟傑偡丅
java -jar $gatk_dir/GenomeAnalysisTK.jar -T UnifiedGenotyper -R $reference -I "s_"$i"_realigned_alnRecal.bam" -nt 4 -L $exon_capture_file -D $dbSNP_file -A AlleleBalance -A DepthOfCoverage -o $outdir/"s_"$i"_SNV.vcf" -stand_call_conf 50.0 -stand_emit_conf 10.0 -dcov 200 -out_mode EMIT_VARIANTS_ONLY
#-L偱椞堟傪巜掕偟偰傗傞偲丄偦偙偵偁傞SNV偩偗弌椡偱偒傑偡丅-stand_call_conf偲-stand_emit_conf偱丄僐乕儖偡傞曄堎丄僼傽僀儖偵揻偒弌偡曄堎偺嵟彫Phred Score傪巜掕丅-dcov偼丄偙傟埲忋偺僇僶儗僢僕偑偁傞偲偒偵丄擖椡偟偨悢偺儕乕僪傪儔儞僟儉偵僒儞僾儕儞僌偟偰丄張棟偵偐偐傞帪娫傪抁弅偟傑偡丅-out_mode偱丄曄堎偺晹暘偩偗弌椡偡傞偐丄慡偰偺墫婎偵偮偄偰弌椡偡傞偐丄側偳偑愝掕偱偒傑偡丅
#懕偄偰丄GATK偺VariantFiltration傪巊偭偰丄SNV偺僼傿儖僞儕儞僌傪峴偄傑偡丅
#傑偢outdir偵堏摦丅
cd $outdir
java -jar $gatk_dir/GenomeAnalysisTK.jar -T VariantFiltration -R $reference -V "s_"$i"_SNV.vcf" -o "s_"$i"_SNV_filtered.vcf" --clusterWindowSize 10 --filterExpression "QUAL < 30.0 || QD < 5.0 || HRun > 5 || SB > -0.10" --filterName "HARD_TO_VALIDATE"
#--clusterWindowSize偼丄SNV偑僋儔僗僞乕偲側偭偰尒偮偐傞乮=杮摉偵偨偔偝傫SNV偑偁傞偺偐傕偟傟側偄偗偳丄False positive偺壜擻惈傕崅偄乯応強傪挷傋傞偨傔偺僂傿儞僪僂僒僀僘傪寛傔傑偡乮偙偙偱偼10墫婎偯偮僂傿儞僪僂傪偢傜偟偰挷傋傞乯丅--filterExpression偱丄vcf僼傽僀儖偺INFO僼傿乕儖僪偵偁傞崁栚偺偆偪丄僼傿儖僞儕儞僌偵巊偆傕偺傪巜掕丅暋悢巜掕偡傞偲偒偼丄||偱嬫愗傝傑偡丅--filterName偱丄filterExpression偛偲偵丄僼傿儖僞乕傪僷僗偟側偐偭偨SNV偵偮偗傞僞僌偺柤慜傪巜掕偱偒傑偡丅
#傑偢outdir/intermediate偵栠傝傑偡丅
cd $outdir/intermediate
#GATK偺UnifiedGenotyper傪巊偭偰丄Indel偺僐乕儖傪偟傑偡丅
java -Xmx2G -jar $gatk_dir/GenomeAnalysisTK.jar -T UnifiedGenotyper -glm INDEL -R $reference -I "s_"$i"_realigned_alnRecal.bam" -nt 4 -L $exon_capture_file -D $dbSNP_file -A AlleleBalance -A DepthOfCoverage -o $outdir/"s_"$i"_indel.vcf" -stand_call_conf 50.0 -stand_emit_conf 10.0 -dcov 200
#-glm乮genotype_likelihoods_model乯偱丄INDEL僐乕儖偱偁傞偙偲傪巜掕偟傑偡丅
#懕偄偰丄Indel偺僼傿儖僞儕儞僌傪峴偄傑偡丅
#傑偢outdir偵堏摦丅
cd $outdir
#GATK偺VariantFiltration傪巊偭偰丄僼傿儖僞儕儞僌傪峴偄傑偡丅
java -jar $gatk_dir/GenomeAnalysisTK.jar -T VariantFiltration -R $reference -V $"s_"$i"_indel.vcf" -o $outdir/"s_"$i"_indel_filtered.vcf" --filterExpression "MQ0 >= 4 && ((MQ0 / (1.0 * DP)) > 0.1)" --filterName "HARD_TO_VALIDATE" --filterExpression "SB >= -1.0" --filterName "StrandBiasFilter" --filterExpression "QUAL < 10" --filterName "QualFilter"
#SNV偺偲偒偲摨偠傛偆偵僷儔儊乕僞乕愝掕丅徻嵶偼丄僿儖僾側偳傕偛嶲徠偔偩偝偄丅
#CombineVariant偱丄SNV偺僼傽僀儖偲Indel偺僼傽僀儖傪傑偲傔傑偡丅
java -Xmx2g -jar $gatk_dir/GenomeAnalysisTK.jar -R $reference -T CombineVariants -V:SNV,vcf $outdir/"s_"$i"_SNV_filtered.vcf" -V:INDEL,vcf $outdir/"s_"$i"_indel_filtered.vcf" -o "s_"$i"_SNV_indel_filtered.vcf"
#snpEff傪巊偭偰丄曄堎偺傾僲僥乕僔儑儞傪峴偄傑偡丅
#僇儗儞僩僨傿儗僋僩儕偵snpEff偺config僼傽僀儖偑懚嵼偟偰偄傞昁梫偑偁傞偺偱堏摦
cd $snpEff_dir
#snpEff偵傛傞傾僲僥乕僔儑儞傪幚峴
java -Xmx4G -jar ./snpEff.jar eff -v -i vcf -o vcf GRCh37.63 $outdir/"s_"$i"_SNV_indel_filtered.vcf" > $outdir/"s_"$i"_SNV_indel_filtered.snpEff.vcf"
#埲忋偱丄傾僲僥乕僔儑儞偮偒偺丄SNV偲Indel傪僐乕儖偟偨vcf僼傽僀儖偑嶌惉偱偒傑偡丅
偲偄偆偙偲偱丄堦捠傝曽朄傪夝愢偝偣偰偄偨偩偒傑偟偨偑丄幚嵺偵惗僨乕僞傪埖偭偰傒側偄偲幚姶偑傢偐側偄丄偲偄偆偺偑惓捈側偲偙傠偩偲巚偄傑偡丅
偱偡偺偱丄僒儞僾儖僨乕僞傪梡偄偨夝愅傪峴側偭偰傒傑偟傚偆丅
惗僨乕僞傪岞奐偟偰偄傞僒僀僩偲偟偰偼丄NCBI偺SRA偑桳柤偱偡偑丄偙偪傜偱偼丄SRA宍幃偺僼傽僀儖偟偐攝晍偝傟偰偍傜偢丄奺帺偱Fastq僼傽僀儖偵曄姺偡傞昁梫偑偁傝傑偡丅
曄姺偼丄SRA Toolkit傪巊偊偽壜擻偱偡偑丄柺搢側応崌偼丄DDBJ乮DNA Data Bank of Japan乯偺DRA偐傜側傜丄Fastq僼傽僀儖傪捈偵僟僂儞儘乕僪偱偒傑偡丅
崱夞偼丄楎惈堚揱偺儊儞僨儖宆堚揱昦偺尨場堚揱巕傪丄椉恊偑嬤恊崶偺徢椺偐傜摼偨DNA偺僄僋僜乕儉夝愅偵傛偭偰摨掕偡傞偙偲傪栚揑偲偟偨尋媶偺丄岞奐偝傟偰偄傞惗僨乕僞傪偲傝偁偘偰傒傑偡丅
尋媶偺榑暥偼偙偪傜
http://www.ncbi.nlm.nih.gov/pubmed/21936905
惗僨乕僞偼偙偪傜
http://trace.ddbj.nig.ac.jp/DRASearch/study?acc=SRP007801
偐傜丄塃懁偺Navigation晹暘偺Experiment棑丄Fastq偺偲偙傠偐傜丄僼傽僀儖偑僟僂儞儘乕僪偱偒傞FTP僒僀僩偵堏摦偱偒傑偡丅
嵽椏偑懙偭偨偲偙傠偱丄忋弎偺僐儅儞僪傪僐僺儁偱擖椡偟偰偄偔偲丄偙傫側姶偠偺僼傽僀儖偑偱偒傞偲巚偄傑偡丅
偙偙傑偱僟僂儞僒僀僘偱偒傞偲丄偁偲偼Excel側偳偱偺夝愅偱傕廫暘懳墳偱偒傞偺偱偡偑丄偙偙偱偼丄annovar偲偄偆僣乕儖傪巊偭偰丄娙扨偵幘姵偺尨場堚揱巕岓曗傪峣傝崬傓曽朄傪徯夘偟偨偄偲巚偄傑偡丅
#椺偺傛偆偵丄嵟怴僶乕僕儑儞偺傕偺傪僟僂儞儘乕僪偟偰丄夝搥偟傑偡丅
#annovar偱偺夝愅傪巒傔傞慜偵丄傑偢丄忋偺夝愅偱弌棃偨vcf僼傽僀儖偐傜丄PASS偺僼儔僌偑偮偄偰偄傞曄堎偩偗傪拪弌偟傑偡丅
grep -e ^# -e PASS $outdir/"s_"$i"_SNV_indel_filtered.snpEff.vcf" > $outdir/"s_"$i"_SNV_indel_filtered.snpEff.PASS.vcf"
#annovar偺僨傿儗僋僩儕偵堏摦偟傑偡丅
cd annovar
#./convert2annovar.pl傪巊偭偰丄vcf宍幃偐傜丄annovar偱夝愅壜擻側宍幃偵曄姺偟傑偡丅
./convert2annovar.pl $outdir/"s_"$i"_SNV_indel_filtered.snpEff.PASS.vcf" -format vcf4 > $outdir/"s_"$i"_SNV_indel_filtered.snpEff.PASS.txt"
#偙偙偱偼丄帺摦偱楎惈堚揱偺儊儞僨儖宆堚揱昦偲壖掕偟偨応崌偺峣傝崬傒傪峴側偭偰偔傟傞丄./auto_annovar.pl傪巊偭偰傒傑偡丅
#僼傿儖僞儕儞僌偺僗僥僢僾偺徻嵶偼丄偙偪傜傪偛嶲徠壓偝偄丅傑偨丄annovar偼丄偙傟埲奜偵傕丄偄傠偄傠偲僇僗僞儅僀僘壜擻側僼傿儖僞儕儞僌朄傪帩偭偰偄傑偡偺偱丄揔媂丄帋偟偰偄偨偩偗傟偽偲巚偄傑偡丅
#傑偢丄夝愅偵昁梫側僨乕僞儀乕僗僼傽僀儖傪僟僂儞儘乕僪偟傑偡丅
./annotate_variation.pl -downdb -buildver hg19 gene humandb
./annotate_variation.pl -downdb -buildver hg19 knownGene humandb
./annotate_variation.pl -downdb -buildver hg19 segdup humandb
./annotate_variation.pl -downdb -buildver hg19 phastConsElements46way humandb
./annotate_variation.pl -downdb -buildver hg19 -webfrom 1000g2010nov humandb
./annotate_variation.pl -downdb -buildver hg19 -webfrom annovar snp130 humandb
#./auto_annovar.pl傪巊偭偰丄曄堎偺峣傝崬傒傪峴偄傑偡丅
./auto_annovar.pl --buildver hg19 -model recessive $outdir/"s_"$i"_SNV_indel_filtered.snpEff.PASS.txt humandb
#嵟廔揑偵惗惉偟偰偒偨僼傽僀儖傪尒傞偲丄20屄偺堚揱巕柤偑巆偭偰偍傝丄偦偺拞偵偼丄尨場堚揱巕偲峫偊傜傟偨SHROOM3傕巆偭偰偄傞偐偲巚偄傑偡丅
偲偄偆姶偠偱丄傂偲捠傝丄僄僋僜乕儉僨乕僞夝愅偺棳傟傪徯夘偟偰傒傑偟偨丅
偨偩丄崱夞偲傝偁偘偨僨乕僞夝愅朄偱丄昁偢忋庤偔偄偔偲偼尷傜側偄偺偑尰幚偱偡丅
椺偊偽丄僒儞僾儖僨乕僞偱偲傝偁偘偨尋媶偱偼丄SNP僠僢僾傪梡偄偨absence-of-heterozygosity region乮儂儌偺曄堎偑楢懕偟偰懕偔椞堟丄嬤恊崶偺椉恊偐傜嫟捠偟偰堚揱偟偨偲峫偊傜傟傞乯偺夝愅傪慻傒崌傢偣偰丄峏側傞峣傝崬傒傪峴側偭偰偄傞偺偱偡偑丄偙偆偄偭偨曽朄傕桳岠偱偡丅
傑偨丄尨場曄堎偑摨掕偱偒側偄棟桼偲偟偰偼丄
丒挷傋偨幘姵偑丄怹摟棪100%偺儊儞僨儖宆堚揱偱側偄丅
丒昞尰宆偺掕媊偑娫堘偭偰偄傞丅
丒尨場曄堎偑僐乕儖偝傟偰偄側偄乮尨場曄堎偑Exome奜偵偁傞応崌傪娷傓乯丅
丒LowQual傗HARD_TO_VARIDATE偵恀偺尨場曄堎偑愽傫偱偄傞丅
丒dbSNP偵搊榐偝傟偄傞偑丄幘姵偺尨場偵側傞曄堎傪岆偭偰徚偟偰偄傞丅
丒庬娫偺曐懚偺掱搙傗梊應僾儘僌儔儉偵傛傞敾掕偱偼廳梫偱側偝偦偆偩偑丄幘姵偺尨場偵側傞曄堎傪岆偭偰徚偟偰偄傞丅
側偳偑峫偊傜傟傞偲巚偄傑偡丅
偝傜偵丄dbSNP偵偮偄偰偺Tips偲偟偰丄
丒dbSNP偵偼丄婛抦偺儊儞僨儖宆堚揱昦偺尨場曄堎傕搊榐偝傟偰偄傞丅
丒dbSNP偼丄130偐傜1000僎僲儉偺僨乕僞偑擖偭偰偒偰偄傞丅
丒1000僎僲儉埲崀偺僨乕僞偵偼丄楎惈偱昦婥偺尨場偲側傞丄昿搙偺掅偄曄堎偑擖偭偰偄傞壜擻惈偑偁傞丅
丒1000僎僲儉埲崀偺僨乕僞偵偼丄FalsePositive傕彮側偔側偄偐傕偟傟側偄丅
偲偄偭偨偙偲傕峫椂偡傞昁梫偑偁傞偲峫偊傜傟傑偡丅
dbSNP135偱偼丄偄偢傟偐偺恖庬偱昿搙偑1%埲忋丄側偳偺婎弨傪枮偨偟偨曄堎偺傒傪娷傓僼傽僀儖偑嶌惉偝傟傞傛偆側偺偱丄婬側幘姵偺尨場堚揱巕傪扵偡応崌丄崱屻偼偙偺僼傽僀儖傪巊偆偺偑椙偄偐傕偟傟傑偣傫丅
偲偄偆傢偗偱丄傑偩傑偩堦嬝撽偱偼偄偒傑偣傫偟丄暋嶨側堚揱條幃傪偲傞偁傝傆傟偨幘姵偺尋媶偱偼丄偝傜偵夝愅朄傪岺晇偡傞昁梫偑偁傝傑偡偑丄僄僋僜乕儉夝愅偑丄堚揱揑塭嬁傪庴偗傞幘姵偺尋媶偵偍偄偰丄旕忢偵桳朷側庤抜偱偁傞偙偲偼娫堘偄側偄偲偙傠偱偟傚偆丅
嵟屻偵丄僄僋僜乕儉夝愅傪梡偄偨幘姵尋媶偵偮偄偰偺丄椙偄Review傪徯夘偟偰偍偒傑偡偺偱丄偛嶲徠壓偝偄丅
Exome sequencing as a tool for Mendelian disease gene discovery
乮Mendelian disease gene discovery偲偁傝傑偡偑丄崱屻偺common disease傊偺墳梡偵偮偄偰傕弎傋偰偁傝傑偡丅乯
Written by 崅揷撃乮mail: atakata@brain.riken.jp乯